Jo Mohan has lived with psoriasis since she was a baby. Now 41, she works full-time and runs a peer support group.... Read more >
Jess Ragusa: Trikafta urgently needed on PBS for cystic fibrosis patients
Jess Ragusa: Trikafta urgently needed on PBS for cystic fibrosis patients
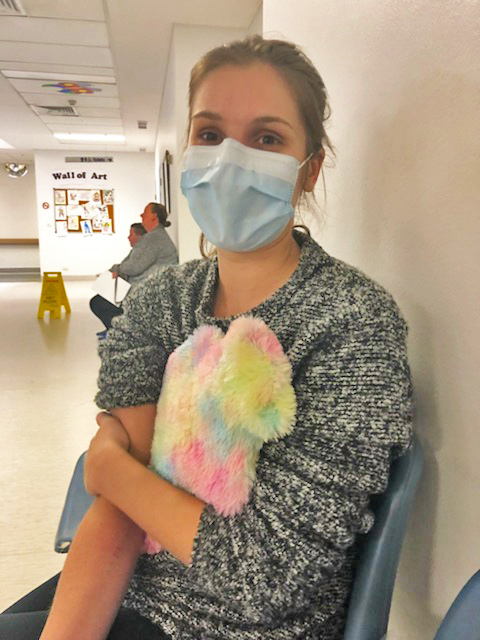
Jess Ragusa’s life has been turned around since she started the new cystic fibrosis (CF) drug, Trikafta. However, it is currently not funded through the Pharmaceutical Benefits Scheme (PBS).
by Rosemary Ainley, 23 March 2022



Trikafta is a revolutionary treatment for cystic fibrosis. It comprises three active ingredients, elexacaftor, tezacaftor and ivacaftor. This is the first time the ingredients have been combined in a triple therapy and studies have shown this approach has significantly improved outcomes for many cystic fibrosis patients.
It is already available in the US and many European countries for CF patients over 12 years and older with at least one copy of the F508del genetic mutation — the most common mutation that results in CF.
There are over 1 million Australians who carry the CF gene and around 3,500 of these have CF. Around 90 per cent of people with CF stand to benefit from Trikafta.
The good news is that Trikafta was recently approved by the Federal Government to be listed on the PBS. However, negotiations between the Federal Government and the manufacturer Vertex Pharmaceuticals (Australia) are yet to be finalised. The cohort of patients who will be eligible is still under discussion.
The problem is, if this process is not concluded before the federal election and before the upcoming retirement of the Minister for Health, Greg Hunt, the last steps that could make Trikafta accessible, may be delayed for many months.
For 27-year-old Jess and other CF patients like her across the country, that may be too long because of the overwhelming financial strain. Their only alternative would be to stop taking Trikafta and revert to managing their CF with less effective treatments.
Jess’s journey and treatments
Jess was diagnosed with cystic fibrosis at birth and commenced medical and physical therapies straight away. She stayed active and fit throughout childhood which helped keep her healthy and meant she only had occasional infections.
At 18, she developed a deep, sticky cough and lost a lot of weight. She was diagnosed with Burkholderia cepacia — a severe antibiotic-resistant infection that greatly reduced her lung function and almost killed her.
Lung function is measured as you breathe out. A healthy range is between 80 and 120 per cent. Jess’s lung function had dropped to around 40 per cent. “With that came the rest of the problems, like fatigue. You’re physically exhausted all the time because breathing is so much harder.”
“They gave me six months to live,” she said. “This bug was going to kill every good cell in my body. They pumped me with IV antibiotics, but it just didn’t work. Nothing could get rid of it. I was given the ‘you fight or you die’ talk. I had just enough energy left to fight.”
Jess worked with a personal trainer every day to help her lungs clear. She also moved from her local hospital to a larger regional hospital to receive more specialised care. “From there, we turned a corner. I got back to ‘ok’ but I was still always sick after that. From someone who never had a hospital admission, I went to three or four a year.”
A change for the worse
Since then, Jess’s life has been hard but she was able to work and never went back to being critically sick. That was until October 2021, when she started coughing up considerable amounts of blood.
“That’s fairly normal for a CF patient, but this one scared me. It was really dark, thick, old blood — which meant it had been sitting there for a long time. But it also had bright red, fresh blood so I knew that I must have had ongoing lung trauma. It burst all the blood vessels in my eyes. My whole face broke out in a bruise. I was swollen all the way down my neck and I had a pounding headache. Never, ever had I had one like that. It was so bad,” she recalled.
Jess called her doctors and was told to go straight to hospital. “I went on IV antibiotics again to try and fight the bug on top of my normal CF stuff for two weeks,” she said. “From that moment, we decided it was time for Trikafta.”
What Trikafta does and how it makes a difference for Jess
Trikafta works by combining the three key ingredients to enhance the structure and activity of the CFTR protein. Tezacaftor and ivacaftor have previously been used to treat CF, however, the addition of elexacaftor is what makes Trikafta superior to earlier treatments.
“This drug has changed my life. I can’t put into words the difference it has made. Within four weeks of starting Trikafta, my lung function went from the 50s to the 80s. I don’t think I’ve been at this level since I was around 10 years old without any scar tissue damage in my lungs. I’m back there,” Jess said. “Now, I don’t cough. I’m off half of my medications because I don’t need to be on them anymore. I have energy. I’ve put on weight. I eat food. I can taste food. I’m sleeping well. For the first time in years, I’m even dreaming. My body is finally able to switch off and relax rather than fighting to keep me alive,” said Jess.
“If you could see how I was and how I am now, it’s like chalk and cheese. You wouldn’t believe the difference a tiny little tablet has made.”

About cystic fibrosis
Cystic fibrosis is usually detected at birth through a heel-prick test or diagnosed in early childhood. In Australia, one in 2,500 babies are born with CF.
It is a genetic condition caused by mutations in the CFTR gene. These mutations affect the structure and activity of the CFTR protein which, in turn, leads to the formation of thick mucus in the lungs, pancreas and gut.
The condition is progressive and leads to premature death, usually due to lung failure. The current average live expectancy of someone with CF is around 35 years, though new treatments like Trikafta are helping to increase life expectancy and quality significantly. There is no cure.
Cystic fibrosis symptoms
Mutations of CF genes cause secretions to become very thick and sticky so they build up and can block the lungs, airways, digestive system and other areas of the body.
A build-up of mucus in the pancreas restricts the flow of enzymes to the small intestine. These enzymes help break down food and allow the nutrients to pass into the bloodstream. Foods that don’t get broken down and absorbed move through the large intestine to be excreted. This means people with CF have a high risk of malnutrition.
Regular infections weaken the immune system and lead to connective tissue scarring which reduces lung elasticity and function. Both the mucus and the scarring make the simple act of breathing a challenge.
Jess’s call for immediate action
Despite her personal challenges, Jess’s motivation for joining forces with Cystic Fibrosis Australia and other CF advocates to get Trikafta onto the PBS is for the other people it will help now and in the future.
“I’m speaking for those who don’t have a voice. The mums who have just found out that their children have CF and are so overwhelmed, as my parents were. They don’t understand what their life is going to look like,” she said. “It may be years before they can access this life-saving medication for their children,” Jess said.
“Trikafta is already helping people come off lung transplant lists. The cost of that alone could far outweigh what the drug costs. Then there’s the impact on the healthcare system. I don’t have to go to the hospital for three or four weeks at a time. Think of how much that is saving the government. Instead, I can work and pay taxes,” she added.
Jess is thankful that some CF patients are close to having access to Trikafta but she is frustrated the process has taken so long. “The Government and Vertex have looked at putting Trikafta on the PBS for several years,” she added. “This time, they’re saying ‘It will happen, we just have to work out when’. I don’t like that answer. I want it NOW. And I want it on the PBS for $6.60 today!”
Jess wants all parties to negotiate, expedite the process and get Trikafta on the PBS before the election. That’s my goal as, once they call the election, they go into caretaker mode and no money gets spent.
Cystic Fibrosis Australia and other key stakeholders recently sent a submission to the Australian Parliament calling for Trikafta to be added to the PBS before the Federal election. The petition received almost 60,000 signatures and will be presented to Parliament soon.
How you can help
“I think it is really important for people to keep talking about cystic fibrosis,” said Jess. “For the first time in years, we were mentioned on TV and mainstream media. People need to understand that CF is the most common life-threatening genetic disease in Australia but people still don’t know about it.
Care about issues like this? Get involved with GHLF Australia
Other ways you can help:
- Donate to the Help Jess have a future GoFundMe campaign.
- Donate to Cystic Fibrosis Australia to help fund their Trikafta Advocacy Plan.
Sources
NPS MedicineWise, Australian Prescriber, articles, Aust Prescr 2021;44:137-8, 2 August 2021, https://www.nps.org.au/australian-prescriber/articles/elexacaftor-tezacaftor-ivacaftor-for-cystic-fibrosis
Cystic Fibrosis Australia, What is CF? https://www.cysticfibrosis.org.au/about-cf/what-is-cf
Standford Medicine, The Cystic Fibrosis Center at Stanford, Education, https://med.stanford.edu/cfcenter/education.html
Australian Government, Department of Health, the Pharmaceutical Benefits Scheme, Public Summary Document – March 2021 PBAC Meeting with May 2021 Addendum, 5.07 elexacaftor/tezacaftor/ivacaftor, https://www.pbs.gov.au/industry/listing/elements/pbac-meetings/psd/2021-05/files/elexacaftor-tezacaftor-ivacaftor-psd-may-2021.pdf
Timothy J Kidd et al, 2008, Burkholderia cepacia complex epidemiology in persons with cystic fibrosis from Australia and New Zealand, NIH, National Library of Medicine, National Center for Biotechnology Information, Res Microbiol, 2008 Apr;159(3):194-9. doi: 10.1016/j.resmic.2008.01.001. Epub 2008 Feb 2, https://pubmed.ncbi.nlm.nih.gov/18356026/
Cystic Fibrosis Australia, Trikafta Advocacy Plan, Communique 382, Petition Update, PRIMETIME, 18 March 2022, https://www.cysticfibrosis.org.au/getmedia/cfdccf1d-af9d-4e77-8ffc-f395e6a69441/Communique_382-Petiton-Update-PRIME-TIME.pdf.aspx
myDr, Tests & Investigations, Lung function tests, https://www.mydr.com.au/lung-function-tests/
Related Posts



This Post Has 0 Comments